Телефон
8 (385) 240-17-84
Адрес:
ул. Малахова, 53, Барнаул
Пн-пт
07:00 - 19:00
Версия для слабовидящих
Духовное развитие
Астрология
Мистика
Нумерология
Религия
Сонник
Христианство
Здоровье
Болезни и условия
Велнесс
Зрение
Психическое здоровье
Рак
Слух
Стоматология
Красота
Волосы
Здоровье кожи лица
Косметика
Уход
Уход за кожей
Лечение зависимостей
Алкоголизм
Курение
Наркомания
Отношения
Брак
Дружба
Знакомства
Неверность
Разрыв отношений
Свадьба
Сексуальность
Самосовершенствование
Вредные привычки
Мотивации
Постановка цели
Психология
Управление стрессом
Самочувствие
Акне
Аллергии
Альтернативная медицина
Болезни
Болезни глаз
Болезни у детей
Вирусные заболевания
Вопросы и ответы
Генетика
Гинекология
Добавки и витамины
Докторская практика
Женское здоровье
Заболевания и лечение
Заболевания ушей
Здоровое питание
Здоровье женщин
Инфекционные заболевания
Кардиология
Лечение
Лечение зубов
Медицина
Мужское здоровье
Неврология
Новообразования
Ногти
Онкология
Отоларингология
Патологии
Патология
Препараты
Прочее
Расстройства
Сон
Травматология
Урология
Спорт и фитнес
Йога
Наращивание мышечной массы
Спорт на открытом воздухе
Физические нагрузки
Снижение веса
Фитнес
Медицина
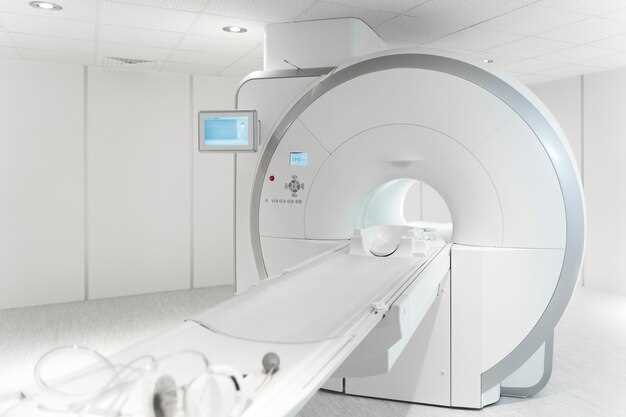
Диагностика лучевая. Методы лучевой диагностики
Подробнее
Что происходит с легкими при курении вейпа
Подробнее
Эффективные методы лечения кашля у грудных детей
Подробнее
Что причиняет появление кровавой рвоты у человека
Подробнее
Эпидемический процесс: характеристика, особенности, фазы
Подробнее
Эмоциональная зрелость: описание, характеристика, способы развития
Подробнее
Здоровье женщин
Эффективные способы удаления папиллом на шее у женщин
Подробнее
Эритроциты понижены у женщины: причины и последствия
Подробнее
Что такое эндометриоз: причины, симптомы, лечение
Подробнее
Что происходит с организмом женщины при бросании курить
Подробнее
Что причиняет боль в правой части живота у женщин
Подробнее
Что принимать при повышенных тромбоцитах в крови у женщин
Подробнее
Женское здоровье
Правда о черных выделениях и их причинах
Подробнее
Эффективные методы лечения насморка у беременных женщин
Подробнее
Эмбрион 5 мм. Какой срок беременности соответствует этому размеру?
Подробнее
Эпизиотомия: описание, внешний вид и лечение швов
Подробнее
Что происходит с внутренними органами при беременности
Подробнее
Что понижает сахар в крови при беременности
Подробнее
Болезни и условия
Нестабильная стенокардия: симптомы и лечение
Подробнее
Нужно знать всем Признаки тромбофлебита, его лечение и профилактика
Подробнее
Причины бронхита, виды, симптомы и лечение у взрослых
Подробнее
Язва двенадцатиперстной кишки: диета, лечение и хирургия | Медицинский портал
Подробнее
Этиология: понятие, примеры, роль в развитии вирусных болезней
Подробнее
Эндометрит: симптомы, лечение, последствия и формы
Подробнее
Лечение
Эозинофилы в анализе крови: расшифровка, норма и отклонения
Подробнее
Что показывает общий клинический анализ мочи
Подробнее
Что показывает бак посев мочи у женщин
Подробнее
Что показывает анализ калограммы у детей
Подробнее
Что пить от частого мочеиспускания: эффективные напитки
Подробнее
Что означает эпителий плоский в анализе мочи женщины
Подробнее
Сейчас читают
Что происходит с легкими при курении вейпа
Подробнее
Что причиняет появление кровавой рвоты у человека
Подробнее
Эффективные методы лечения кашля у грудных детей
Подробнее
Эпидемический процесс: характеристика, особенности, фазы
Подробнее
Диагностика лучевая. Методы лучевой диагностики
Подробнее
Эффективные способы удаления папиллом на шее у женщин
Подробнее
1
2
3
…
104
Вперед »